

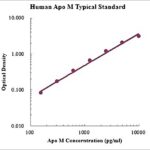
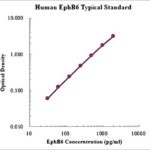
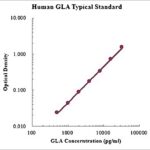
This assay employs the quantitative sandwich enzyme immunoassay technique for the quantitative detection of human GLA. The Human alpha-Galactosidase A/GLA ELISA is for research use only. Not for diagnostic or therapeutic procedures.


Related Products





Recently Viewed Product
-
BioOcean® ELISA KITS, Human Elisa Kit
Human Fas Ligand ELISA Kit
$529.00Quick ViewHuman Fas Ligand ELISA Kit
This assay employs the quantitative sandwich enzyme immunoassay technique for the quantitative detection of Human Fas Ligand. The Human Fas Ligand ELISA is for research use only. Not for diagnostic or therapeutic procedures.
$529.00 -
BioOcean® ELISA KITS, Mouse Elisa Kit
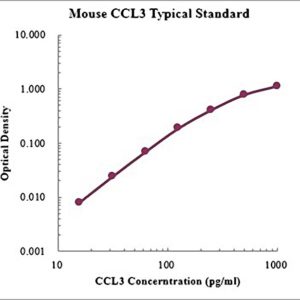
CCL3 / MIP-1α ELISA Kit, Mouse
$529.00Quick ViewCCL3 / MIP-1α ELISA Kit, Mouse
This assay employs the quantitative sandwich enzyme immunoassay technique for the quantitative detection of mouse CCL3. The Mouse CCL3/MIP-1α ELISA is for research use only. Not for diagnostic or therapeutic procedures.
$529.00 -
 BioOcean® ELISA KITS, Mouse Elisa Kit
BioOcean® ELISA KITS, Mouse Elisa KitAXL ELISA Kit, Mouse
$529.00Quick ViewAXL ELISA Kit, Mouse
This assay employs the quantitative sandwich enzyme immunoassay technique for the quantitative detection of mouse AXL. The Mouse AXL ELISA is for research use only. Not for diagnostic or therapeutic procedures.
$529.00 -
BioOcean® ELISA KITS
IFN-γ ELISA Kit, Rat
$529.00Quick ViewIFN-γ ELISA Kit, Rat
This assay employs the quantitative sandwich enzyme immunoassay technique for the quantitative detection of rat IFN-γ. The Rat IFN-γ ELISA is for research use only. Not for diagnostic or therapeutic procedures.
$529.00

{kind=link}
{kind=link}
Reviews
There are no reviews yet.